
Myotonien und Myotone Dystrophien
Klinische Symptomatik
Unter dem Begriff „Myotonie“ versteht man eine Relaxationsstörung oder Dekontraktionshemmung der Willkürinnervation der Muskulatur. Der Patient empfindet dies als Muskelsteifigkeit. Der Myotonie liegt eine heterogene primäre Erkrankung des Muskels im Sinne einer Muskelkanalerkrankung zugrunde. Unterschieden werden dabei
Nicht-dystrophe Myotonien:
- Chlorid-Kanalmyotonien (Typ Thomson, Becker)
- Calcium-Kanalmyotonien
- Natriumkanalmyotonien
und Dystrophische Myotonien:
- Myotone Dystrophie Typ 1 (DM1, Curschmann-Steinert)
- Myotone Dystrophie Typ 2 (DM2, PROMM-Syndrome, Ricker disease)
Die Differenzierung zwischen nicht-dystropher und dystropher Myotone erfolgt zunächst klinisch. Bei den nicht-dystrophen Myotonien liegen in der Regel neben der Myotonie nur eine Kreatinkinaseerhöhung und keine weiteren Symptome vor. Zumeist ist bei diesen Erkrankungen die Muskulatur eher kräftig entwickelt. Wenn Symptome wie Muskelschwäche und Muskelatrophie mit Myotonie im Vordergrund stehen, liegt in der Regel eine dystrophe Myotonie vor, die dann einer ergänzenden Analyse bedarf. Da es sich bei den dystrophen Myotonien um sog. Multisystemerkrankungen handelt, findet man häufig weitere Krankheitssymptome wie Kreatinkinase- und Leberenzymerhöhungen, diabetogene Stoffwechsellage, und einen grauen Star (eine Katarakt). Nicht-dystrophe Myotonien werden sowohl dominant als auch rezessiv vererbt, während dystrophe Myotonien immer dominant vererbt werden.
Klinische Synopsis Myotoner Dystrophien
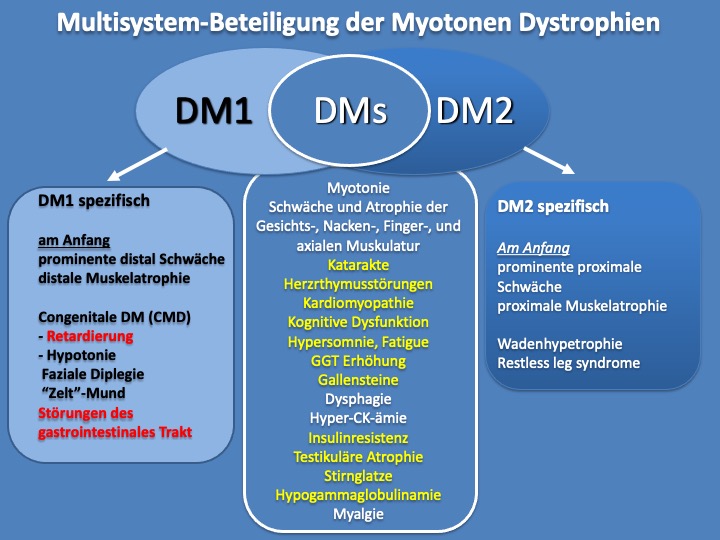
Diagnostik
Im ersten Schritt einer Abklärung sollte eine Krankengeschichte mit Familienstammbaum erhoben und eine körperliche Untersuchung durchgeführt werden. Da aber am Beginn der Erkrankung möglicherweise nur isolierte Symptome auftreten, bleibt die klinische Diagnose oft schwierig. Hier kann durch Zusatzuntersuchungen wie die Elektromyographie (EMG) eine weitere Bestätigung der Erkrankung erfolgen (Nachweis der Myotonie im EMG). Ist das Anfangssymptom z.B. ein grauer Star (Katarakt) oder eine Herzrhythmusstörung wird der Verdacht möglicherweise durch einen anderen Facharzt (Augenarzt oder z.B. Internist) ausgesprochen, bevor dann der Neurologe die endgültige Diagnose stellt.
Heute kann durch eine Blutuntersuchung (Gentest) die Diagnose für myotone Erkrankungen gestellt werden. Der Nachweis der Gen-Veränderung (Mutation) ist beweisend für eine DM1 und 2, sowie für Chlorid-, Calcium- und Natriumkanalerkrankungen.
Die Muskelbiopsie ist in der erweiterten Differentialdiagnose oder im Rahmen wissenschaftlicher Studien sinnvoll.
Therapiemöglichkeiten
Eine ursächliche Therapie wie z.B. eine Gentherapie myotoner Erkrankungen ist bisher noch nicht möglich. Krankengymnastik und eine am Symptom orientierte Behandlung mit dem Ziel die Beeinträchtigung zu mildern ist aber möglich. Zur Stabilisierung der muskulären Schwäche kann zusätzlich Kreatin eingenommen werden. Sollte eine ausgeprägte Myotonie vorliegen, ist eine medikamentöse Therapie (zum Teil sog. off-Label use) z.B. mit Mextiletin, oder Lamotrigin möglich. Engmaschige EKG- und Laborkontrollen sind zu beachten!
Für die Behandlung der Herzsymptome stehen alle üblichen internistisch-kardiologischen Möglichkeiten zur Verfügung (z.B. Defibrillator-Schrittmacher). Eine Blutzuckererkrankung oder Schilddrüsenfunktionsstörung sollte nach üblichen internistischen Regeln konsequent behandelt werden. Durch eine heute zumeist ambulant durchführbare Operation und Einbringung einer künstlichen Linse wird ein Katarakt beseitigt. Graduiertes körperliches Training, Physiotherapie, Ergotherapie und Logopädie sind wichtige Pfeiler der Therapie. Eine kognitive Verhaltenstherapie ist bei Fatigue indiziert. Eine humangenetische Beratung ist immer indiziert, insbesondere wegen der Möglichkeit des Auftretens einer schweren congenitalen myotonen Dystrophie.
Forschung und klinische Studien
Die Forschungsprojekte des Instituts zu Myotonien und myotonen Dystrophien umfassen erweiterter Phänotyp-Korrelationen, sowie klinische Studien zur symptomatischen Therapie und molekulare Grundlage und neue Therapienoptionen (siehe die Forschungsprojekte).
Ansprechpartner im Institut
Prof. Dr. Benedikt Schoser
PD Dr. Stephan Wenninger
Dr. Federica Montagnese
Dr. Natalia Garcia Angarita
Zuletzt geändert: Februar 2022