
Immunzellen produzieren Atherosklerose-förderndes Signalmolekül
München, 01.06.2011
Atherosklerose – umgangssprachlich auch als Gefäßverkalkung bekannt – gehört in der westlichen Welt zu den häufigsten Todesursachen: Chronische Entzündungen in den Blutgefäßen führen zu Ablagerungen, die die Gefäße verengen und letztendlich Herzinfarkte und Schlaganfälle auslösen können. Verursacht wird die chronische Entzündung durch eine außer Kontrolle geratene Reaktion des Immunsystems. Sogenannte dendritische Zellen spielen für die körpereigene Abwehr eine zentrale Rolle, indem sie andere Abwehrzellen aktivieren. Die genaue Funktion dendritischer Zellen bei der Entstehung einer Atherosklerose war bisher allerdings noch unklar. Ein internationales Team von Wissenschaftlern um Professor Christian Weber von der Ludwig-Maximilians-Universität (LMU) München und Privatdozentin Alma Zernecke von der Universität Würzburg konnte nun zeigen, dass dendritische Zellen mithilfe des Signalmoleküls CCL17 einen Selbstregulierungsmechanismus des Immunsystems unterdrücken, der die Immunreaktion abschwächt oder begrenzt – die Entzündung wird zum Dauerzustand. Aber die Wissenschaftler sind bereits einem potenziellen Gegenmittel auf der Spur: „Mit einem Antikörper gegen CCL17 konnten wir das Fortschreiten der Atherosklerose verhindern“, berichtet Weber. CCL17 bietet somit auch die Chance für neue therapeutische Ansätze. (Journal of Clinical Investigation, 1. Juni 2011)
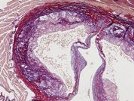
Atherosklerose kann sich unterschiedlich manifestieren: Besonders gefährlich wird es, wenn sich atherosklerotische Ablagerungen lösen und Gefäße im Herz oder im Gehirn verstopfen – dann sind Herzinfarkte und Schlaganfälle die Folge. Verengte Gefäße können aber auch zu Durchblutungsstörungen in den Beinen oder zu der sogenannten vaskulären Demenz im Gehirn führen. Ursache der Atherosklerose sind Schädigungen der inneren Gefäßwand, die zu einer chronischen Entzündung führen: Aus dem Blut wandern Zellen des Immunsystems an die geschädigte Stelle und produzieren Signalstoffe, die weitere Immunzellen zum Ort des Geschehens rufen. Schließlich bilden sich atherosklerotische Plaques, die stetig neue Signalstoffe aussenden, bis die Immunantwort entgleist.
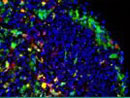
Zu diesem Zweck nutzten die Wissenschaftler transgene Mäuse, deren dendritische Zellen anstelle des Gens für CCL17 mit der genetischen Information für das Grün-fluoreszendierende Protein (GFP) ausgestattet sind, und damit dessen Produktion und den Aufenthaltsort der Zellen nachvollziehbar machen. Auf diesem Weg konnten die Wissenschaftler nicht nur untersuchen, wie sich fehlendes CCL17 auswirkt, sondern auch mithilfe mikroskopischer Methoden – insbesondere der Multiphotonen-Mikroskopie – beobachten, wie dendritische Zellen in den atherosklerotischen Plaques akkumulieren und mit T-Zellen interagieren.
In weiteren Experimenten wurden Mäuse mit T-Zellen aus CCL17-defizienten Mäusen rekonstituiert und umgekehrt in CCL17-defizienten Mäusen regulatorische T-Zellen entfernt. „Als Ergebnis dieser Untersuchungen zeigte sich, dass CCL17 die Atherosklerose vorantreibt, indem es einen wichtigen Schutzmechanismus der Immunantwort unterdrückt“, erklärt Weber: Normalerweise wird die Immunreaktion durch regulatorische T-Zellen (Tregs) gebremst, die die Aktivität anderer Immunzellen hemmen und so dafür sorgen, dass das Immunsystem im Gleichgewicht bleibt. Nun konnten die Wissenschaftler nachweisen, dass in Anwesenheit von CCL17 weniger Tregs in dem entzündeten Gewebe aktiv sind – deren protektiver Mechanismus wird durch CCL17 somit ausgeschaltet.
Publikation:
„CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T-cell homeostasis in mice”;
C. Weber, S. Meiler, Y. Döring, M. Koch, M. Drechsler, R.T.A. Megens, Z. Rowinska, K. Bidzhekov, C. Fecher, E. Ribechini, M.A.M.J. van Zandvoort, C.J. Binder, I. Jelinek, M. Hristov, L. Boon, S. Jung, T. Korn, M.B. Lutz, I. Förster, M. Zenke, T. Hieronymus, T. Junt, A. Zernecke;
Journal of Clinical Investigation, 2011;121(7): 2898-2910.
doi:10.1172/JCI44925
Abstract
Immune mechanisms are known to control the pathogenesis of atherosclerosis. However, the exact role of DCs, which are essential for priming of immune responses, remains elusive. We have shown here that the DC-derived chemokine CCL17 is present in advanced human and mouse atherosclerosis and that CCL17+ DCs accumulate in atherosclerotic lesions. In atherosclerosis-prone mice, Ccl17 deficiency entailed a reduction of atherosclerosis, which was dependent on Tregs. Expression of CCL17 by DCs limited the expansion of Tregs by restricting their maintenance and precipitated atherosclerosis in a mechanism conferred by T cells. Conversely, a blocking antibody specific for CCL17 expanded Tregs and reduced atheroprogression. Our data identify DC-derived CCL17 as a central regulator of Treg homeostasis, implicate DCs and their effector functions in atherogenesis, and suggest that CCL17 might be a target for vascular therapy.