
Myotone Dystrophien
Hintergrund
Die Erstbeschreibungen der klassischen Myotonen Dystrophie (Typ 1) 1909 gehen in Deutschland auf Steinert und in den USA auf Batten und Gibb zurück (DM1, OMIM 160900 und 602668). 1912 entdeckte Curschmann die familiäre Häufung der Katarakte, so dass er 1936 den Begriff der Multisystem-Erkrankung einführte. Diese klassische Form stellt die häufigste Muskelerkrankung des Erwachsenenalters dar. In Ergänzung zur Muskeldystrophie und Myotonie werden häufig zusätzliche Symptome wie Herzrhythmusstörungen, posteriore subkapsuläre Katarakte und endokrine Störungen (u.a. Diabetes mellitus) gefunden. Viele detaillierte klinische Beschreibungen zeigen, dass für die DM1 eine variable Penetranz, eine Antizipation (früherer Beginn und schwererer Verlauf in der Folge-Generation) und ein bei maternaler Transmission erhöhtes Risiko für eine schwere kongenitale Form nachweisbar war und diese Aspekte nicht den einfachen Mendelschen Regeln unterliegen. 1992 konnte die genetische Ursache der DM1 identifiziert werden. Es wurde ein abnorm expandiertes CTG Triplettrepeat im 3'-UTR (untranslated region) des Dystrophia myotonica Proteinkinase-Gens (DMPK) auf Chromosom 19 gefunden. Normalerweise sind bei Gesunden bis 37 CTG Kopien vorhanden, bei DM1 Patienten zwischen 50 und 4000 CTG Kopien. Die Länge der CTG-Repeat-Expansion korreliert mit dem Beginn und der Schwere der klinischen Manifestation. 1994 wurde primär in Deutschland durch Professor Kenneth Ricker, und parallel in den USA durch Professor Charles Thornton, eine zweite multisystemische myotone Dystrophie beschrieben. Bei diesen Familien stand eine proximal betonte Schwäche zum Beispiel beim Treppensteigen oder aus dem Stuhl-Hochkommen im Vordergrund, so dass Ricker das Akronym PROMM (Proximale Myotone Myopathie) wählte. Laura Ranum lokalisierte 1998 das Krankheits-Gen auf Chromosom 3q. 2001 gelang dann die Identifikation eines abnorm expandierten Tetranukleotid CCTG repeat im Intron 1 des Zinkfinger-9 Gens (ZNF-9) auf Chromosom 3q bei deutschen und amerikanischen PROMM Familien. Inzwischen sind alle unter PROMM oder mit anderen Akronymen beschriebene Familien weltweit als Myotone Dystrophie Typ 2 (DM2, OMIM 116955) genetisch klassifiziert. Bei Gesunden liegen bis 27 CCTG Kopien vor und bei DM2 Patienten zwischen 75 und mehr als 11.000 CCTG Kopien. Bei der DM2 konnte keine eindeutige Antizipation oder Korrelation der CCTG-Repeat-Expansions-Länge zur Schwere der Manifestation nachgewiesen werden. Darüber hinaus ist eine kongenitale Form der DM2 nicht beschrieben.
Molekulare RNA-Pathogenese der myotonen Dystrophien
Trotz zumindest zweier verschiedener Genloci der myotonen Dystrophien scheint pathogenetisch ein gemeinsamer Mechanismus vorzuliegen: die abnormen repeat Expansionen, transkribiert in die prä-mRNA, akkumulieren in sog. ribonukleären Foci oder Inklusionen im Zellkern und Zytoplasma. Die langen CUG oder CCUG repeat Stränge können Sekundärstrukturen bilden (z.B. sog. Haarnadelformationen) und Proteine des Zellkernes binden oder beeinflussen. Wahrscheinlich wird dadurch besonders der Spleißapparat und Wechselwirkungen von Transkriptionsfaktoren mit spezifischen DNA-Bindungsstellen gestört (Folge: sog. aberrantes Spleißen).
Alternatives Spleißen kommt bei 40-60% der Gene des humanen Genoms vor. Pre-mRNAs können in multiple Proteinisoformen mit unterschiedlichen Funktionen und/oder gewebsspezifischer und entwicklungsspezifischer Expression übersetzt werden. Alternatives Spleißen stellt einen zusätzlich regulatorischen Mechanismus dar, durch den gewebespezifische und entwicklungszeit-spezifische Isoformen von Proteinen in Zellen exprimiert werden können. Somit führt alternatives Spleißen zu einer weiteren Dimension der Komplexität auf Proteinebene ohne die Anzahl der kodierenden Gene in komplexen Organismen zu erhöhen. Eine Depletion und Redistribution von Chromatin- assoziierten Transkriptionsfaktoren, RNA-Bindungsproteinen wie Muscleblind like Protein, CELF Proteine oder CUG und CCUG-Bindungsproteinen, die das alternative splicing auf bisher nicht genau bekannte Weise regulieren, binden an Sequenz-spezifische Abschnitte der pre-mRNA um ein alternatives Exon ein- oder auszuschleusen (Inklusion - Exklusion) und akkumulieren in sog. ribonuklearen Foci im Zellkern und Zytoplasma (Abbildung 1). MBNL1 und CELF Proteine scheinen antagonistische RNA-Regulatoren zu sein. Fehlgesteuertes alternatives Spleißen führt zu organspezifischen Auswirkungen, deren molekulare Grundlagen partiell für einige multisystemischen Symptome der DMs verstanden sind.
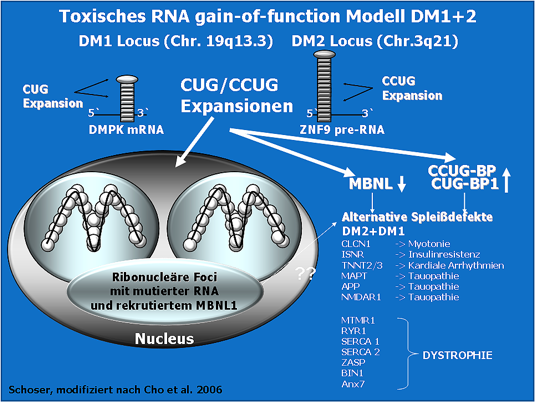
Toxisches RNA gain-of-function Modell DM1+2 (Schoser, modifiziert nach Cho et al. 2006)
Ziele
Im Vordergrund der eigenen Forschung steht die Weitentwicklung der Phänotypbeschreibung (assoziierte Symptome, Epidemiologie), die klinische Forschung für die Versorgung der Betroffenen und die Molekularpathogenese der DM2.
Kooperationspartner
- In unserer deutschen DM2-Arbeitsgruppe (mit Dr. Kress, Würzburg, PD Dr. Schneider-Gold, Bochum, PD Dr. Kornblum, Bonn) beschäftigt wir uns als klinische Forschergruppe mit klinischen Fragestellungen
- Mit Professor Ranum, Gainsville, University of Florida, USA werden Untersuchungen zur mRNA ZNF9 Expression und grundsätzlichen Repeat-Mechnismen durchgeführt. (MDA-grant, Co-Investigator)
- Mit Professor Timchenko, University of Houston, USA werden Untersuchungen zur Charakterisierung von CCUG und CUG-Bindungsproteinen durchgeführt (MDA-grant, Co-Investigator)
Ansprechpartner
Prof Dr. med. Benedikt Schoser
Ausgewählte Publikationen
-
Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. (Neuromuscul Disord. 2011 Jun;21(6):443-50. Epub 2011 May 4.)Udd B, Meola G, Krahe R, Wansink DG, Bassez G, Kress W, Schoser B, Moxley R.
-
Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. (Nat Med. 2011 Jun;17(6):720-5. Epub 2011 May 29.)Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, de Munain AL, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N.
-
Non-ATG Initiated Translation Directed by Microsatellite Expansions (Proc Natl Acad Sci U S A. 2011 Jan 4;108(1):260-5. Epub 2010 Dec 20.)Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP.
-
Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. (Curr Genomics. 2010 Apr;11(2):77-90.)Schoser B, Timchenko L.
-
Myotone Dystrophien (Akt. Neurol 37:348-359, 2010)Schneider-Gold C, Grimm T, Kress W, Schoser B
-
Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. (Eur J Neurol. 2010 Jun 1;17(6):842-5. Epub 2010 Jan 20.)Kirzinger L, Schmidt A, Kornblum C, Schneider-Gold C, Kress W, Schoser B.
-
Reduction of the rate of protein translation in patients with myotonic dystrophy 2. (J Neurosci. 2009 Jul 15;29(28):9042-9.)Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L.
-
Myotonic Dystrophies type 1 and 2 – a summary of current aspects. (Semin Pediatr Neurol. 2006 Jun;13(2):71-9.)Schara U, Schoser BG.