
Myofibrilläre Myopathien
Myofibrilläre Myopathien (MFM) und Gliedergürteldystrophien (LGMD) bilden eine klinisch und genetisch heterogene Gruppe genetisch determinierter, progredienter Erkrankungen des Skelettmuskels, teilweise auch mit Beteiligung des Herzmuskels. Gemeinsames Symptom ist eine fortschreitende Muskelschwäche und –atrophie, die häufig erst im Erwachsenenalter eintritt und sich in Verteilungsmuster und Schweregrad zwischen den verschiedenen Formen deutlich unterscheidet. Bislang wurden LGMD und MFM im Wesentlichen histologisch voneinander unterschieden. Mit fortschreitender Identifizierung von Loci und Genen für LGMD und MFM entstehen jedoch derzeit neue Zuordnungen und Klassifikationen, sowie ein verbessertes Verständnis der Pathogenese. So wurden interessanterweise identische Mutationen des Myotilin-Gens sowohl mit der LGMD1A als auch mit einer MFM in Verbindung gebracht.
Morphologisch werden myofibrilläre Myopathien durch anormale, intrazelluläre, desminpositive Proteinaggregate charakterisiert, die sich in den Muskelfasern befinden. Bei Desminopathien handelt es sich um eine genetisch definierte Subgruppe der Myofibrillären Myopathien, welche durch Mutationen im Desmin-Gen auf Chromosom 2q35 verursacht werden. Diese Erkrankungen wurden früher primäre Desminopathien oder Desmin-Aggregationsmyopathien genannt, da es zu einer Akkumulation von Desmin in den Muskelfasern kommt. Des Weiteren konnten in Patienten mit MFM Mutationen in anderen Genen identifiziert werden, die entweder für Komponenten der Z-Scheiben kodieren oder daran beteiligt sind, deren Struktur aufrecht zu erhalten: Myotilin (MYOT, TTID), ZASP (LDB3), Filamin C (FLNC) und alpha-B-Crystallin (CRYAB), four and a half LIM domain 1 (FHL1), Bcl-2-associated athanogene-3 (BAG3) und Plectin (PLEC1).
Die klinischen Phänotypen Myofibrillärer Myopathien, die durch Mutationen im Desmin-Gen verursacht werden, erstrecken sich über ein weites Spektrum. Innerhalb einer Familie treten scapuloperoneale, Gliedergürtel- und distal betonte Verteilungsmuster der Muskelschwäche auf mit variierender kardialer oder respiratorischer Beteiligung. Auch reine Kardiomyopathien sind bekannt. Die meisten Patienten zeigen ein autosomal-dominantes Vererbungsmuster. Es sind allerdings sowohl Patienten mit einer autosomal-rezessiven Vererbung, als auch Patienten ohne ersichtliche familiäre Vorgeschichte dokumentiert worden. Desmin ist ein 53 kDa schweres, muskelspezifisches Intermediärfilament-Protein der Skelett-, Herz- und glatten Muskulatur. Es stellt eine wichtige, strukturelle Komponente der muskulären Zytoarchitektur dar und bildet ein drei-dimensionales Gerüst rund um die myofibrillären Z-Scheiben. Es verbindet den gesamten kontraktilen Apparat mit dem subsarcolemmalen Zytoskelett, dem Zellkern und anderen Organellen. Das pathologische Merkmal Myofibrillärer Myopathien sind Desminaggregate in den Muskelfasern betroffener Patienten. Obwohl die meisten der bisher identifizierten krankheits-verursachenden Mutationen im Desmin-Gen ein Intermediärfilament-Gerüst bilden, wenn sie in vitro oder in Zellkultur analysiert werden, werden alle Mutationen mit der Bildung dieser Desminaggregate in Zusammenhang gebracht.
Molekulare Analyse und Entwicklung molekularer Therapien für myofibrilläre Myopathien - neue Erkenntnisse über die Auswirkungen verschiedener Mutationen in myofibrillären Genen auf die Protein-Assemblierung mittels konfokaler Einzelmolekülspektroskopie
Auch wenn die Akkumulation von aggregiertem Desmin einen einheitlichen histopathologischen Befund darstellt, muss immer noch aufgeklärt werden, wie die verschiedenen, identifizierten Mutationen auf molekularer Ebene bei den betroffenen Patienten zu Fehlfunktionen der Muskelzellen führen. Ein besseres Verständnis dieser molekularen Signalwege könnte zu neuen Therapieansätzen führen, da bis heute nicht geklärt ist, ob eine Verbesserung der physiologischen Filamentbildung oder aber eine Reduktion der pathologischen Aggregate wichtiger ist, um eine normale Zellfunktion aufrecht zu erhalten. Die Tatsache, dass die Desmin-Assemblierung auf unterschiedliche Weise durch die einzelnen Mutationen beeinflusst wird und diese zu unterschiedlichen Phänotypen (z.B. vorwiegende Beteiligung der Skelettmuskulatur versus vorwiegende kardiale Beteiligung) führen, zeigt, dass verschiedene Signalwege vorhanden sein müssen.??Um die Desmin-Aggregation quantitativ auf Einzelmolekülebene zu analysieren, verwenden wir Fluoreszenz-Korrelations-Spektroskopie (FCS), Fluorescence Intensity Distribution Analysis (FIDA) und Scanning for Intensely Fluorescent Targets (SIFT) in einem konfokalen Einzelmolekül-Detektionssystem. In den letzten Jahren stellte sich heraus, dass sich diese Messmethoden hervorragend dazu eignen, die Assemblierungs-Kinetiken fluoreszenzmarkierter Proteine in Echtzeit und die molekulare Zusammensetzung von Proteinaggregaten zu bestimmen. Des Weiteren können so viele Hochdurchsatz-Messungen mit kleinen Probenvolumina durchgeführt und neue Substanzen für Therapieansätze gefunden werden.
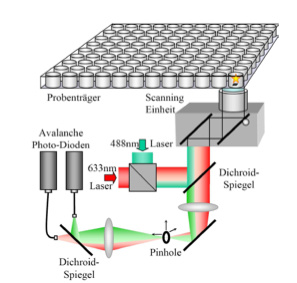
Confocal single molecule detection system
Diese Technik ermöglicht es, die veränderten Assemblierungseigenschaften der Desmin-Mutanten zu detektieren und deren Auswirkungen auf Einzelmolekülebene zu untersuchen. Diese Effekte sind mittels klassischer Fluoreszenzmikroskopie nicht messbar. Mittels SIFT-Messungen werden Protein-Protein Interaktionen analysiert und die Effekte der pathogenen Mutationen charakterisiert. Der Einsatz dieser Technologie bietet die molekulare Basis für eine detaillierte funktionelle Klassifikation der Mutationen im Desmin-Gen und kann dazu beitragen, neue Diagnosemöglichkeiten und Therapieansätze zu entwickeln.
Charakterisierung der pathologischen Proteinaggregate durch Laser-Mikrodissektions-Mikroskopie und Proteom-Analyse.
Ziel dieses Forschungsvorhaben ist die Identifikation und Charakterisierung der four and a half LIM domain Gen (FHL1) assoziierter Myopathien mit Analyse der Skelettmuskelpathologie mit Hilfe der Laser-Mikrodissektions-Mikroskopie der Inklusions- und Aggregat-Pathologie und anschließender Proteom-Analyse im Vergleich zu bekannten und bis dato unbekannten Proteinaggregat-Myopathien. Dies stellt einen alternativen Ansatz zum Verständnis humaner Proteinaggregations-Myopathien dar.
FHL1-Gen assoziierte Myopathien (FHL1opathien) umfassen bis dato die Reducing Body Myopathy (RBM), die X-chromosomale Myopathie mit posturaler Muskelatrophie (XMPMA), die Emery-Dreifuss Muscular Dystrophy (EDMD) und die scapuloperoneale Myopathie (X-SM). Alle vier Myopathien sind seltene progressive Erkrankung der Muskulatur und u.a. charakterisiert durch spezifische Muskelfasereinschlüsse, sog. zytoplasmatische und reduzierende Körperchen. In Ihrer schwersten Form als RBM führen sie noch im Kindesalter zum Tod. Alle vier Erkrankungen werden durch Mutationen in unterschiedlichen Abschnitten des FHL1 Gens verursacht. Die bisher bekannte Muskelpathologie der FHL1opathien entspricht neben den spezifischen Einschlusskörperchen der einer myofibrillären Myopathie mit ganulofilamentärem Material und Z-Linienveränderungen.
Im Rahmen dieses Forschungsprojekts sollen weiterer Patienten und Familien aus dem bisher noch unbekannten Spektrum der FHL1opathie mit deren klinischer, histopathologischer und genetischer Charakterisierung identifiziert werden. Zudem werden Proteom-Spektren mit Hilfe der Laserdissektions-Mikroskopie Technik aus Einschlusskörperchen und Aggregaten von genetisch klassifizierten myofibrillären Myopathien für Vergleichsstudien evaluiert und spezifische Aggregat-Bestandteile mit Hilfe der Proteom-Analyse und ggf. anschließender Genuntersuchung von bisher unklassifizierten myofibrillären Myopathien analysiert. Schließlich liefert der Einsatz dieser Technik auch optimiertes Material für die spezifische Analyse von humanem und nicht-humanem Untersuchungsmaterial.
Drittmittelförderung
Diese Forschungsprojekte werden im Rahmen einer DFG Forschergruppe FOR1228 durch die Deutsche Forschungsgemeinschaft (DFG) gefördert.
Kooperationspartner
- Prof. Dr. Rolf Schröder, Erlangen, Deutschland
- PD Dr. Christoph Clemen, Köln, Deutschland
- Prof. Dr. Dieter Fürst, Bonn, Deutschland
- Prof. Dr. Matthias Vorgerd, Bochum, Deutschland
- PD Dr. Ludwig Eichinger, Köln, Deutschland
- Prof. Dr. Harald Herrmann-Lerdon, Heidelberg, Deutschland
- Prof. Dr. Gerhard Wiche, Wien, Österreich
- Prof. Dr. Franz-Georg Hanisch, Köln, Deutschland
Ansprechpartner
Ausgewählte Publikationen
-
Divergent effects of pathogenic mutations in primary desminopathy on oligomer assembly and multimerization of desmin revealed by single particle spectroscopy. (JNEN 2010;69:415-424)Levin J, Bulst S, Thirion C, Schmidt F, Bötzel K, Krause S, Pertl C, Kretzschmar H, Walter MC, Giese A, Lochmüller H.
-
The p.G154S mutation of the alpha-B crystallin gene (CRYAB) causes late-onset distal myopathy (Neuromuscul Disord. 2010 Apr;20(4):255-9. Epub 2010 Feb 19.)Reilich P, Schoser B, Schramm N, Krause S, Schessl J, Kress W, Müller-Höcker J, Walter MC, Lochmuller H.
-
Myofibrillar myopathies: a clinical and myopathological guide. (Brain Pathol. 2009 Jul;19(3):483-92.)Schröder R, Schoser B.
-
Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. (Brain. 2009 Feb;132(Pt 2):452-64. Epub 2009 Jan 29.)Schessl J, Taratuto AL, Sewry C, Battini R, Chin SS, Maiti B, Dubrovsky AL, Erro MG, Espada G, Robertella M, Saccoliti M, Olmos P, Bridges LR, Standring P, Hu Y, Zou Y, Swoboda KJ, Scavina M, Goebel HH, Mitchell CA, Flanigan KM, Muntoni F, Bönnemann CG.
-
Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. (Neuromuscul Disord. 2008 Jun;18(6):475-82. Epub 2008 May 27.)Strach K, Sommer T, Grohé C, Meyer C, Fischer D, Walter MC, Vorgerd M, Reilich P, Bär H, Reimann J, Reuner U, Germing A, Goebel HH, Lochmüller H, Wintersperger B, Schröder R.
-
Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. (J Clin Invest. 2008 Mar;118(3):904-12)Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, Sewry C, Battini R, Hu Y, Cottle DL, Rosenblatt M, Spruce L, Ganguly A, Kirschner J, Judkins AR, Golden JA, Goebel HH, Muntoni F, Flanigan KM, Mitchell CA, Bönnemann CG.
-
An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. (Am J Hum Genet. 2008 Jan;82(1):88-99.)Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, Lohberger B, Farra N, Petek E, Schwarzbraun T, Ofner L, Löscher WN, Wagner K, Lochmüller H, Vincent JB, Quasthoff S.
-
Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. (Brain. 2007 Dec;130(Pt 12):3250-64.)Kley RA, Hellenbroich Y, van der Ven PF, Fürst DO, Huebner A, Bruchertseifer V, Peters SA, Heyer CM, Kirschner J, Schröder R, Fischer D, Müller K, Tolksdorf K, Eger K, Germing A, Brodherr T, Reum C, Walter MC, Lochmüller H, Ketelsen UP, Vorgerd M.
-
Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. (Brain. 2007 Jun;130(Pt 6):1485-96. Epub 2007 Apr 17.)Walter MC, Reilich P, Huebner A, Fischer D, Schröder R, Vorgerd M, Kress W, Born C, Schoser BG, Krause KH, Klutzny U, Bulst S, Frey JR, Lochmüller H.